
Resumen
El síndrome de Sotos o gigantismo cerebral se caracteriza por macrocefalia, hipercrecimiento, retraso mental y anomalías en el sistema nervioso central. Pueden presentar defectos congénitos cardiacos. Se presenta el caso de un paciente con síndrome de Sotos y sus alteraciones a nivel bucodental. Se realiza su tratamiento dental con anestesia general.

Dra. M» Cristina Moreno Martín.
Odontóloga.
Master en Rehabilitación Oral Implantosoportada de ESORIB.
Práctica privada.
D. José Manuel de los R’os de la Pe–a
Enfermero de la USBD-D del Sermas.
Dra. Maitena Urberuaga Erce
Médico-Odontoestomatóloga.
Servicio Vasco de Salud. Osakidetza.
D.» Rosa Domingo-Malvad’.
Enfermera.
Servicio Vasco de Salud. Osakidetza.
Palabras clave
Síndrome de Sotos, gigantismo cerebral, macrocefalia, retraso mental.
Abstract
Sotos syndrome or cerebral gigantism is characterized by macrocephaly, overgrowth, mental retardation, and central nervous system abnormalities. Congenital heart defects may be present. We introduce a Sotos syndrome patient and the dental findings. The oral treatment is made under general anaesthesia.
Key words
Sotos syndrome, cerebral gigantism, macrocephaly, mental retardation.
Introducción
El síndrome fue descrito en 1964 por Sotos y colaboradores. También lo denominaron gigantismo cerebral porque había un crecimiento excesivo y rápido durante los primeros 4 años de vida, con peso, talla y edad ósea avanzados para su edad cronológica. Después el crecimiento continúa paralelo al percentil 97 o mayor. La circunferencia de la cabeza ha sido bien documentada por encima del percentil 98 (1).
Los pacientes tienen un peso de nacimiento promedio de 3.400 gr, lo que implica unos percentiles de 75 a 90. La talla de nacimiento promedio es de 55,2 cm, mayor del percentil 97. La edad ósea adelantada con respecto a la edad cronológica, en muchos casos es a expensas de una mayor maduración falángica que de los huesos del carpo. Las manos y pies pueden ser grandes en comparación con el resto del cuerpo y los pies planos o desviados hacia dentro, pueden ser frecuentes.
El hallazgo clínico fundamental es el hipercrecimiento prenatal y postnatal. La talla adulta en varones se sitúa en torno a 184,3+-6,0 cm y en mujeres en torno a 172,9+- 5,7 cm y se han descrito tallas adultas más elevadas (1, 2).
Algunos tienen un coeficiente de inteligencia normal pero la mayoría (80-85%) tienen deficiencia mental. Las manifestaciones del sistema nervioso central son frecuentes. El retraso en las adquisiciones del desarrollo, para caminar, hablar y del lenguaje, están casi siempre presentes y la torpeza es frecuente (60-80%) por hipotonía y laxitud articular (1). Aparece disfunción neurológica no progresiva con dilatación del sistema ventricular compatible con hidrocefalia comunicante y a veces alteraciones sugerentes de atrofia cerebral (3) con espacios subaracnoideos aumentados que habitualmente no requieren tratamiento (4).
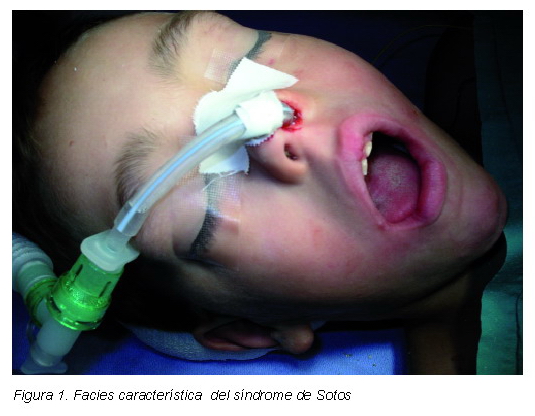
La configuración craneofacial es muy característica. Aparece cabeza grande con un cráneo alto y estrecho o macro-dolicocefalia y frente prominente, hipertelorismo, oblicuidad antimongoloide de los párpados, nariz con puente plano, barbilla afilada, frente prominente y disminución de la línea del cabello frontoparietal (5) (Figura 1).
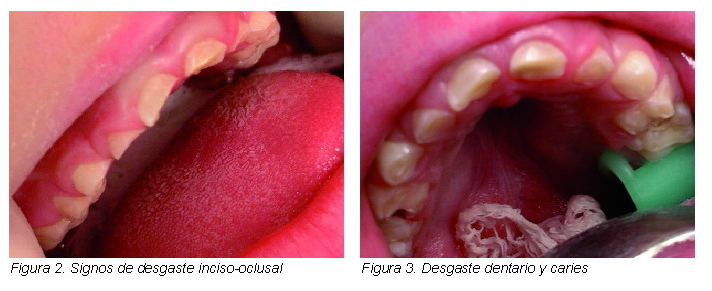
A nivel dental hay una temprana erupción de los dientes (alrededor de los tres meses) que aparece en alrededor del 60-80% de los pacientes (1), babeo prolongado y suelen tener respiración bucal. La mandíbula suele ser prominente. Aparecen con mayor frecuencia signos de desgaste dentario (Figuras 2 y 3).
Pueden tener asociadas alteraciones del comportamiento como fobias, agresiones, obsesiones, adherencia a la rutina, conductas de tipo autista y trastorno de hiperactividad (6, 7).
Aparecen convulsiones en el 30% de los casos y una incidencia incrementada de tumores cerebrales (2,2-3,9%). Pueden aparecer también defectos septales cardíacos, cifoescoliosis, alteraciones urogenitales, como hidronefrosis, riñones hipoplásicos, agenesia renal y dilatación de la pelvis renal, hernias inguinales, alteraciones oftalmológicas, como estrabismo y anomalías del nervio óptico o de la retina y alteraciones endocrinas del tipo de hipotiroidismo primario entre otras, pero ninguna que explique el rápido crecimiento de estos pacientes (1, 4).
La etiología es desconocida y el diagnóstico se basa en los datos somatométricos y los rasgos fenotípicos peculiares ya que los marcadores bioquímicos y endocrinos son normales (8). Las manifestaciones clínicas más características son la configuración craneofacial y el crecimiento excesivo. Sin estos datos no puede realizarse el diagnóstico. Se propugna que hay una causa genética y que existe una herencia autosómico dominante pero la mayoría de los casos en la actualidad son esporádicos. Recientemente Kurotaki et al (9) han descrito mutaciones en el gen NSD1 (5q35) en el 60-75% de los casos esporádicos, indicando que la haploinsuficiencia del gen NSD1 es la causa más importante del síndrome de Sotos (10, 11).
Caso clínico
Se presenta el caso de un niño de 6 años y un mes de edad con retraso madurativo generalizado y diagnóstico de gigantismo cerebral. Nacido a término, de parto eutócico con un peso de 3.900 gr. Entre los hábitos parafuncionales aparecen bruxismo, onicofagia, respiración bucal y babeo persistente.
A la exploración intraoral se encuentran: caries en molares temporales, desgaste oclusal en dentición decidua, mordida abierta anterior y paladar con tendencia ojival. Las características físicas concuerdan con el diagnóstico de síndrome de Sotos (Figuras 1-3).
 |
Se realiza el tratamiento con anestesia general por la nula colaboración del paciente, consistente en selladores oclusales en molares definitivos, pulpotomías en 8.4 y 7.4, obturaciones en el resto de molares temporales que lo precisan, tartrectomía y flúor tópico.
 |
Se realiza una revisión de control postquirúrgico a la semana y luego a los seis meses encontrando como hallazgos la erupción de 1.1 y 2.1 sin exfoliación de los temporales, así como una fístula en 7.4. Se plantean exodoncias que son llevadas a cabo en consulta ambulatoria y con anestesia local ya que se ha conseguido mejorar el manejo de la conducta del paciente.
Discusión
El aspecto craneofacial y el crecimiento excesivo del paciente concuerdan con el diagnóstico de síndrome de Sotos. Por la familia se tiene conocimiento de una erupción dentaria prematura, alrededor de los tres meses. También aparecen babeo prolongado y respiración bucal, así como una mandíbula prominente y paladar ojival.
La afectación de los huesos craneofaciales favorece la presencia de malposiciones que junto con los problemas de conducta en estos pacientes y las dificultades para una dieta adecuada, así como una higiene correcta, favorecen la presencia de caries.
El comportamiento de tipo autista hizo necesario el tratamiento con anestesia general, pero el manejo de la conducta fue mejorando y en las revisiones posteriores se pudieron realizar exodoncias en consulta sin que hasta la fecha haya sido necesario acudir a una nueva anestesia general.
La educación para la salud es una herramienta fundamental para contribuir a la mejora de la salud bucodental de las personas con discapacidad capacitando a sus cuidadores en el cuidado. Al disminuir la incidencia de patología, consecuentemente diminuye la necesidad de tener que realizar un nuevo tratamiento bucodental con anestesia general (12) minimizando así los riesgos inherentes a la misma, ya que es un procedimiento que no está exento de riesgos.
Correspondencia
Carmen Martín Sanjuán. Avda. de Buenos Aires, 97, 7. d. 20018 Madrid.
Teléfono: 913032965
Correo electrónico: cmsanjuan7@yahoo.es.
Bibliografía
1. Sotos JF. Actualización del hipercrecimiento. An Pediatr (Barcelona). 2004; 60: 79-86.
2. Rial JM, Rodr’guez I, Gonz‡lez JP, L—pez R. Diagnóstico diferencial del hipercrecimiento. BSCP Can Ped 2001; 26(1): 1-9.
3. Funez F, Talesnik E, Muzzo S. Gigantismo cerebral o síndrome de Sotos. Rev Chil Pediatr 1979; 50(3): 61-64.
4. Gusmao D, Xavier A, Almeida MA, Monteiro J, Vieira JD, Santos AC. Sotos syndrome (cerebral gigantism): analysis of 8 cases. Arq Neuro-psiquiatr 2002; 60(2A): 234-238.
5. Alves J, Cury V, Leonezi E. Síndrome de Sotos: um estudo de caso. Rev Uniara 2005; 16: 227-234.
6. Mulas F, Hern‡ndez-Muela S, Etchepareborda MC, Abad-Mas L. Bases clínicas neuropediátricas y patogénicas del trastorno del espectro autista. Rev Neurol 2004; 38(Supl 1): S9-S14.
7. Stella L. Avances en neurobiología del autismo. Acta Neurol Colomb 2006; 22: 91-96.
8. Bravo M, Chac—n J, Bautista E, PŽrez-Camacho I, Trujillo A, Grande MA. Síndrome de Sotos asociado a distonía focal. Rev Neurol 1999; 28: 971-972.
9. Kurotaki N, Imaizumi K, Harada N et al. Haploinsufficiency of NSD1 causes Sotos syndrome. Nat Genet 2002; 30: 365-366.
10. R’o M, Clech L, Amiel J et al. Spectrum of NSD1 mutations in Sotos and Weaver syndromes. J Med Genet 2003; 40: 436-40.
11. Visser R, Matsumoto N. Genetics of Sotos syndrome. Current Opinion Pediatr 2003; 15(6): 598-606.
12. Carracedo-Cabaleiro ME, Mart’n-Sanju‡n C, Urberuaga-Erce M, Domingo-Malvad’ R, GutiŽrrez-Crespo C, Moreno-Mart’n MC. Proyecto de Educación para la Salud de la Unidad de Salud Bucodental para personas con discapacidad de 6 a 18 años del Servicio Madrileño de Salud. E. Arch Odontoestomatol 2007; 23(2): 82-88.


